Alpha-1 Antitrypsin-Mangel: Was schützt die einen – und die anderen nicht?
Forschende am MPI für Biochemie haben mithilfe des räumlichen Deep Visual Proteomics-Workflows herausgefunden, warum einige Patienten mit der Erbkrankheit Alpha-1-Antitrypsinmangel trotz des genetischen Defekts gesund bleiben.
Forschende der Abteilung für Proteomik und Signaltransduktion von Matthias Mann am Max-Planck-Institut für Biochemie in Martinsried bei München haben im Rahmen einer internationalen Studie Lebergewebe von deutschen und dänischen Patienten analysiert. Dabei kam das Proteomanalyse-Verfahren Deep Visual Proteomics zum Einsatz. Nun ist klar, warum manche Patienten mit der erblichen Erkrankung Alpha-1 Antitrypsin-Mangel trotz Gendefekts gesund bleiben. Die Ergebnisse der Studie wurden im Fachmagazin Nature veröffentlicht.

Auf den Punkt gebracht:
- Erbliche Erkrankung Alpha-1-Antitrypsin-Mangel: Forschende haben herausgefunden, warum manche Patienten trotz eines Gendefekts gesund bleiben, während andere eine schwere Leberfibrose entwickeln
- Deep Visual Proteomics: verwendete Methode kombiniert KI-Bildanalyse und Massenspektrometrie um unterschiedlicher Proteinzusammensetzung in Leberzellen zu analysieren
- Proteinaggregate in Leberzellen: Krümelförmige Aggregate sind frühe Gegenreaktion gestresster Zellen auf Erkrankung; ballförmige Aggregate treten später auf und korrelieren mit der Leberfibrose
- Hoffnung auf klinische Anwendung: Ein möglicher Ansatz wäre die Entwicklung eines Leberfibrose-Frühwarnsystems, das auf der frühen peroxisomalen Antwort der Leberzellen basiert.

In diesem Video erklärt Florian Rosenberger, Erstautor der Veröffentlichung, den Ablauf der Studie. (in englisch)
Das Protein Alpha-1-Antitrypsin ist ein Enzymhemmer, ein Proteinasen-Inhibitor. Gebildet wird er in der Leber, aber er wirkt in der Lunge. Dort hat er die Aufgabe, die Produktion von Immunzellen zu regulieren, da eine zu starke Reaktion der Immunzellen eine Lungenerkrankung verursachen kann. Jedoch haben manche Menschen einen Gendefekt, der bewirkt, dass sich die Alpha-1-Proteine nicht korrekt falten. Die Leber produziert dann zu wenig Alpha-1 – und somit erreichen auch zu wenige Enzymhemmer die Lunge. Die genetische Mutation, die Ursache des Mangels ist, wird entweder von der Mutter oder vom Vater oder von beiden ans Kind vererbt.
Die heterozygote Form, bei der nur ein Elternteil den Gendefekt weitergibt, hat in Europa eine von 20 Personen. Viele merken oft ihr ganzes Leben lang nichts davon oder werden nur leicht krank. Um einiges gefährlicher ist die seltenere homozygote Form, bei der der Gendefekt sowohl vom Vater als auch von der Mutter stammt. Davon ist eine von 2000 Personen betroffen. Die Wahrscheinlichkeit einer Erkrankung ist hier sehr viel höher. Und die Erkrankungen sind auch schwerwiegender – wobei nicht nur die Lunge etwa in Form einer chronisch obstruktiven Lungenerkrankung (COPD) betroffen sein kann, sondern auch die Leber selbst: Die Menschen entwickeln dann zum Beispiel eine schwere Leberfibrose oder Tumore.
Gleiche Mutation, unterschiedliche Krankheitsverläufe
Zur homozygoten Form des Gendefekts hat eine internationale Studie unter der Leitung des Max-Planck-Instituts für Biochemie in Martinsried bei München nun neue wichtige Erkenntnisse hervorgebracht. Erstautor Florian Rosenberger von der Abteilung für Proteomik und Signaltransduktion erklärt, worum es geht: „Bei der homozygoten Form ist eine Sache auffällig. Da es sich um eine monogenetische Erkrankung handelt und alle Patienten die gleiche Mutation haben, müsste es theoretisch immer die gleichen Krankheitsverläufe geben. Das ist aber nicht der Fall: Ein Drittel der homozygoten Patienten entwickelt eine schwere Leberfibrose, eine Ablagerung von Bindegewebe in der Leber, die die Leberfunktion einschränkt. Zwei Dritteln bleiben dagegen gesund. Wir wollten herausfinden, was dahintersteckt und welche molekularen Mechanismen bestimmen, dass einige Menschen mit homozygotem Alpha-1-Mangel gesund bleiben und andere nicht.“
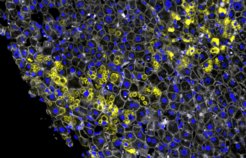
Das Verfahren, mit dem diese Frage geklärt werden konnte, heißt Deep Visual Proteomics. Es ist in Martinsried und Kopenhagen in den Proteomik-Gruppen von Matthias Mann entwickelt worden und darauf spezialisiert, mit Hilfe von Proteomanalysen die tiefliegenden Ursachen von Erkrankungen aufzuspüren. Grundlage der Alpha-1-Studie waren die Lebergewebeproben von deutschen und dänischen Leberfibrose-Patienten. Florian Rosenberger weiter: „Wir haben uns Proben über das komplette Krankheitsspektrum hinweg angeschaut. In den frühen Stadien – also dann, wenn die Erkrankung noch nicht klinisch auffällig ist – konnten wir sehen, wie es dem Körper gelingt, die beginnende Krankheit doch noch aufzuhalten.“ Für die Bildanalyse der Mikroskopieaufnahmen benutzte das Forscherteam ein spezielles künstliches neuronales Netz, ein sogenanntes Convolutional Neural Network (CNN). Es war vorher darauf trainiert worden, menschliche Gesichter und Alltagsgegenstände zu erkennen.
Auf die Feinheiten kommt es an
In der Studie zeigte sich, dass das CNN auch mit menschlichen Leberzellen sehr gut zurechtkommt und in der Lage ist, unterschiedliche äußerliche Merkmale der Erkrankung in der Zelle auseinanderzuhalten. Konkret ging es um Anhäufungen des Alpha-1-Proteins in den Hepatozyten, eine Folge des Krankheitsausbruchs. „Unser CNN konnte feinste Unterschiede in den Morphologien dieser Aggregate erkennen“, so Rosenberger. Am auffälligsten waren zwei Erscheinungsformen. Krümelförmige Aggregate mit einer eher rauen amorphen Struktur. Und ballförmige Aggregate mit einer definierteren Struktur. Die Frage lautete: Was unterscheidet die Zellen in diesen beiden Zuständen? Sind die unterschiedlichen Formen Zufall oder steckt ein Mechanismus dahinter?
Hier gelang Rosenberger und dem Team ein Durchbruch. Sie konnten die molekularen Ereignisse – Bildung der Proteinkrümel oder -bälle sowie die Übergangsphasen vom einen zum anderen – in die richtige zeitliche Abfolge bringen und so herausbekommen, was die unterschiedlichen Formen der Aggregate bedeuten. Die krümelförmige Morphologie tritt zuerst auf. Es ist eine sehr frühe Gegenreaktion der gestressten Zelle auf die Erkrankung, sichtbar in speziellen Zellorganellen, den Peroxisomen. Die ballförmige Morphologie tritt dagegen später auf, wenn die Leberfibrose schon weit fortgeschritten ist. Wobei das Zellstadium nicht mit der Schwere der Erkrankung korreliert, so Rosenberger. Die ballförmigen Aggregate finden sich auch in Patienten mit niedriggradiger Fibrose. „Die Veränderung vom Krümel zum Ball ist ein Kernpunkt der Studie. Sie zeigt, welche kompensatorischen Mechanismen in den Leberzellen in welcher Reihenfolge ablaufen. Wie die Zellen versuchen, die Aggregatbildung und damit die Leberfibrose zu bekämpfen.“
Auf dem Weg zur klinischen Anwendung
Bei der Aufklärung dieser Mechanismen spielte die verbesserte KI-basierte Bildanalyse eine entscheidende Rolle. „Die jüngsten technologischen Fortschritte in der Massenspektrometrie waren entscheidend“, sagt Professor Matthias Mann. „Wir können jetzt Einzelzellmessungen durchführen und so detaillierte molekulare Informationen aus einer kleinen Menge Gewebe, sogar aus einzelnen erkrankten Leberzellen, extrahieren.“
Die Ergebnisse der Studie könnten schon bald klinische Relevanz erlangen. Die Entwicklung einer Fibrose bei Menschen mit der homozygoten Mutation könnte möglicherweise verhindert werden. „Bei der Auswertung der Krankengeschichten haben wir festgestellt, dass bei Personen mit schwerer Fibrose die frühe peroxisomale Reaktion fehlt“, erklärt Rosenberger. „Wir wissen jetzt, dass diese Reaktion schützend wirkt. Unser Ziel ist es, ein Frühwarnsystem für Leberfibrose zu entwickeln – eine Möglichkeit, Risikopatienten zu identifizieren, bevor Symptome auftreten.“
Aleksander Krag, Professor an der Universität von Süddänemark und Leiter des Odense Liver Research Center, betont: „Indem wir die Alpha-1-Antitrypsin-Akkumulation auf Einzelzellebene gemessen haben, konnten wir frühe molekulare Auslöser für das Fortschreiten des Alpha-1-Antitrypsin-Mangels identifizieren. Das könnte die Grundlage für die Verbesseerung von Therapien für Patienten sein."
Professor Pavel Strnad, Hepatologe am Universitätsklinikum Aachen und langjähriger Kooperationspartner des Projekts, ergänzt: „Eine fehlerhafte Proteinfaltung ist für viele menschliche Erkrankungen, darunter Parkinson und Alzheimer, von zentraler Bedeutung. Die Untersuchung einer monogenetischen Erkrankung wie den Alpha-1-Antitrypsin-Mangel bietet eine einzigartige Gelegenheit, den Krankheitsverlauf besser zu verstehen. Diese Arbeit vertieft unser Verständnis von Proteinfaltungsstörungen und ihren Folgen. Dies wird auch über den Alpha-1-Antitrypsin-Mangel hinaus von Bedeutung sein.“
Die Studie wurde in der Fachzeitschrift Nature veröffentlicht.
Glossar:
Deep Visual Proteomics: ist eine Methode die 2022 in der Abteilung Mann entwickelt wurde. Die Methode kombiniert moderne Mikroskopie, maschinelles Lernen, Lasermikrodissektion und ultra-sensitive Massenspektrometrie mit nachfolgenden bioinformatischen Analysen (Mund et al., Nature Biotechnology, 2022).
Enzyme: sind Proteine, die als Katalysatoren tätig sind. Das heißt, dass ihr mitwirken biologische Reaktionen beschleunigt.
Massenspektrometrie: ist eine analytische Technik, die Ionen nach ihrem Masse-zu-Ladung-Verhältnis trennt und misst, um chemische Substanzen oder Moleküle zu identifizieren und zu quantifizieren. Es handelt sich um eine grundlegende Technologie in der Proteomik, die die Identifizierung und Quantifizierung tausender Proteine in komplexen biologischen Proben ermöglicht.
Proteasen-Inhibitor: ist eine Substanz, die die Aktivität von eiweißspaltenden Enzymen, sogenannten Proteasen, hemmt, um den Abbau von Proteinen zu verhindern.
Proteom: umfasst die Gesamtheit aller Proteine in einem Lebewesen, einem Gewebe oder einer Zelle zu einem bestimmten Zeitpunkt. Das Proteom ist hoch dynamisch und reagiert auf die Anforderungen der Zelle, sowie auf Krankheiten oder Umwelteinflüsse.
Proteomik: ist die Erforschung des Proteoms











